Research, current (for examples of previous work, scroll down)
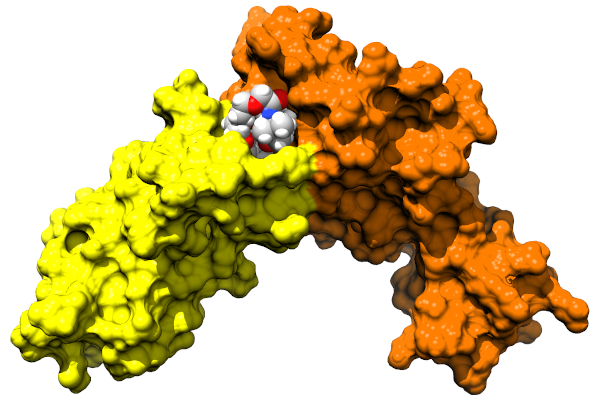
Molecular degraders are compounds that can trigger targeted protein degradation in cells. They are an alternative to traditional small molecule inhibibtors, with a number of advantages. At Celeris Therapeutics we use a combination of machine learning/artificial intelligence and molecular simulation based approaches, to develop Proteolysis Targeting Chimeric Molecules (PROTAC) as novel drug candidates for a range of targets.
Previous research
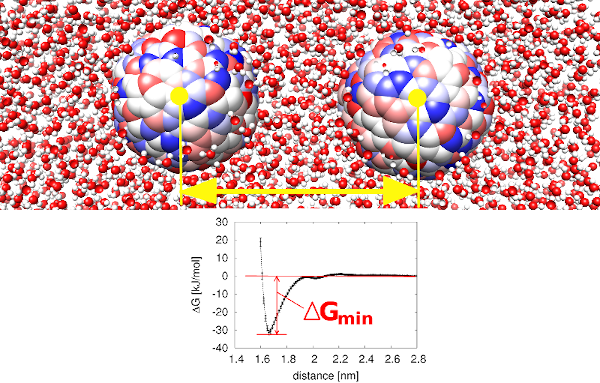
Aggregation of proteins in concentrated solutions can be a major issue in formulation and manufacturing of biopharmaceuticals. In a study, using molecular simulation and simple model systems, we established the relative importance of structure based descriptors for predicting the stability of protein solutions (poster).
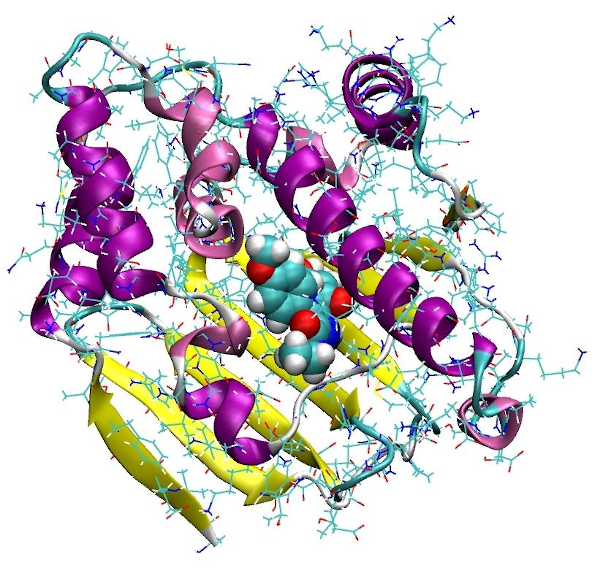
Hsp90 is an interesting cancer target. We developed an in-silico pipeline for efficient screening of Hsp90 inhibitors. (conference presentation)
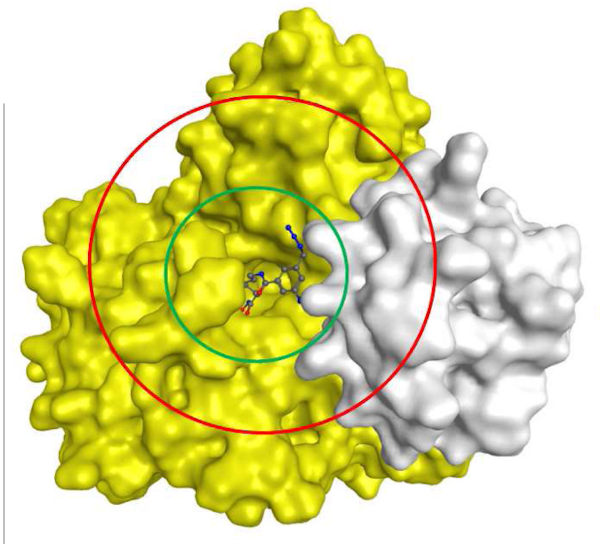
Histone deacetylases are a group of proteins involved in the regulation of gene expression. For this target, I employed a number of computational approaches, including multiple receptor docking the prediction of ligand binding pose details, and protein-complex structures, to assist the interpretation of SAR, towards improved affiniy and selectivity.
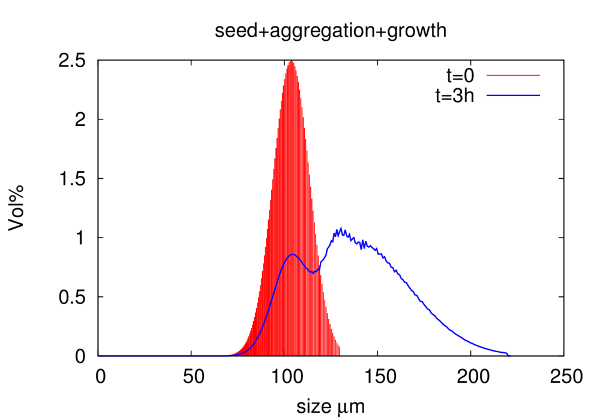
Crystallization can be used as an efficient technique for purification in manufacturing of bio-pharmaceuticals. We developed and implemented a fast solver for population balance equatons, accounting for nucleation, growth, and aggregation, to model this process in-silico. In combination with FBRM measurements the tool can be applied to optimize process parameters of anti-solvent crystallization of protein solutions.
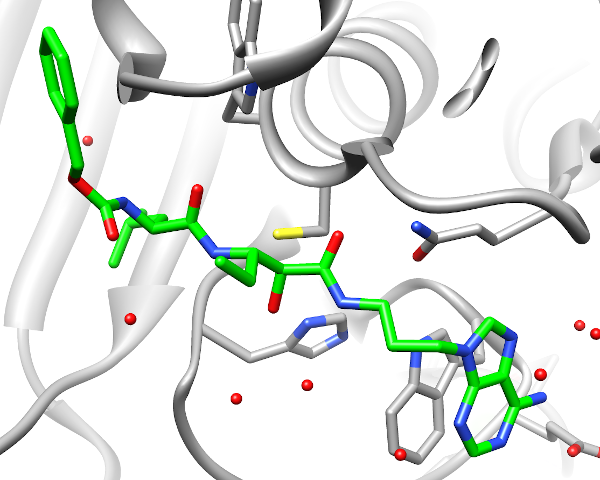
Calpain, a cystein-protease, is a potential target for a number of diseases. Common inhibitors for this target include molecules that are covalently bound to the proteins active cysteine. We developed a methodology for virtual screening of covalent inhibitors, combining the transition state geometry, and a geometrical analysis of docking poses.
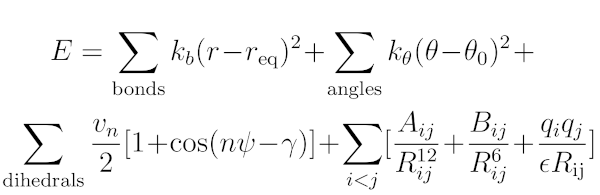
An important part of any molecular simulation approach is the force field/model potential, employed to calculate energies and forces, based on a molecular conformation and configuration. I designed a number of tools to facilitate and accelerate the force filed development. The results can be used for various applications, including the simulation of small molecules and synthetic polymers (see here).
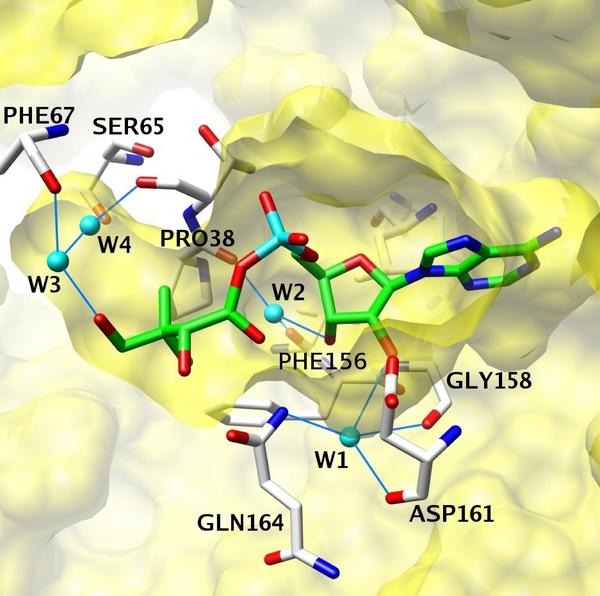
Pantothenate Synthetase, a TB target, features a number of conserved water molecules in its binding site. As part of a virtual screening campaign, we used MD based free energy calculations to estimate the relative stability of each water molecule. The results were used and to identify candidates that can be replaced by inhibitors to improve binding affinities, (poster)
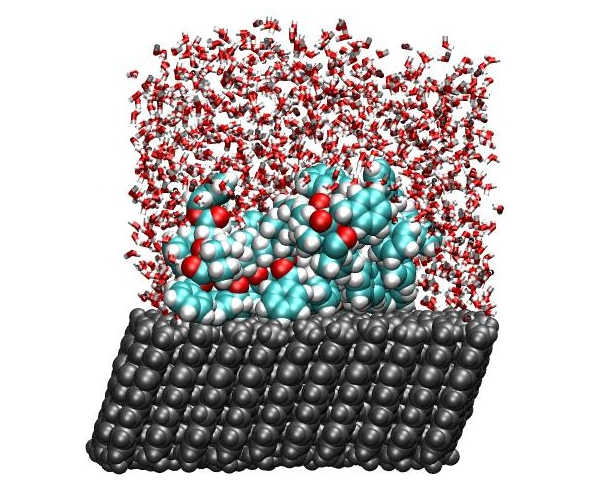
Block-co-polymers can be used as efficient stabilizers in colloidal solutions, e.g., pigment based ink-jet inks. To accelerate the development of such inks we developed a simulation based method to quantify the interactions between polymers and pigment surfaces in-silico (presentation)
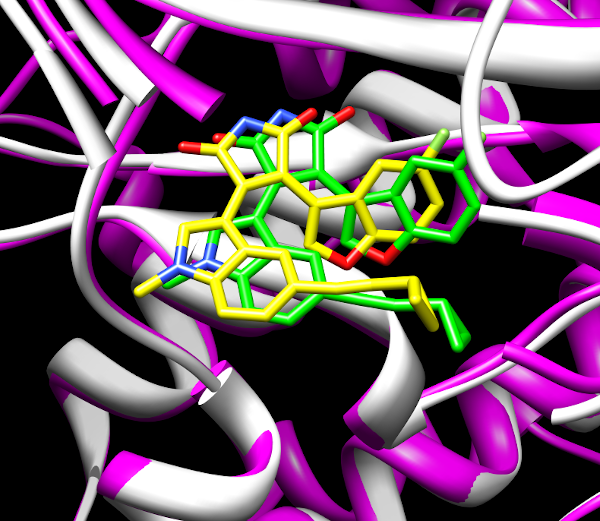
The scaffold of a common type of kinase inhibitors results in a pronounced symmetry of the ligand/receptor interactions. A comparison with crystal structures demonstrates that simple docking/scoring approaches frequently misrepresent the orientation of these inhibitors in the binding site. Using MD simulations and LIE/MMPSSA based analysis we developed a method to reliably predict binding poses.
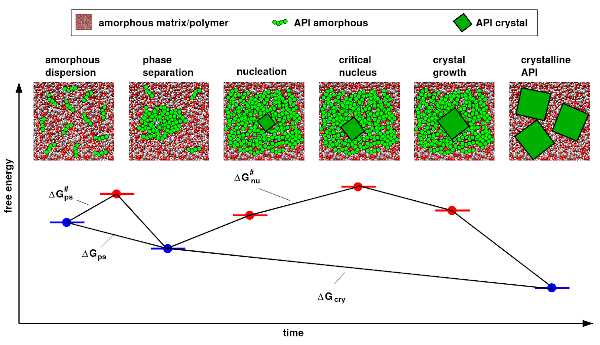
A strategy to improve the bio-availibility of poorly soluble small molecule drugs is their formulation as amorphous solid dispersion (ASD). We performed study to better understand the contributions of kinetics (mobility) versus energetics (solubility) of the drug in a polymer matrix, towards in-silico prediction of relative ASD stabilities. The resulting MD based workflow can be used accelerate the development of ASD based formulations, by screening different polymer types for a given drug candidate.
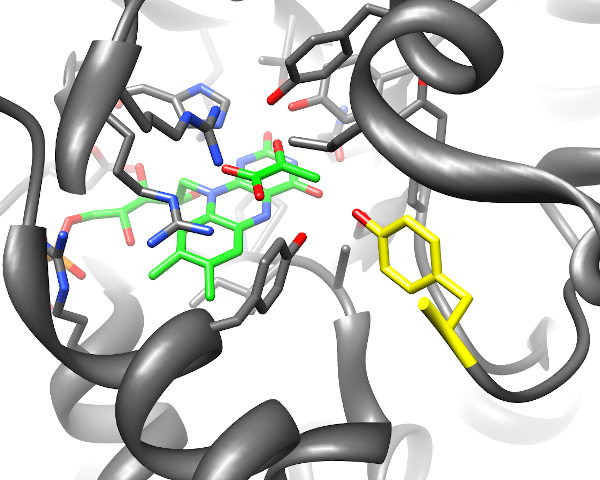
Enzyme reaction mechanisms: Using MD simulations I contributed to elucidating details of substrate binding and reaction kinetics of enzymes with potential applications in bio-technology, including lactat-oxidase, Mannitol-2-Dehydrogenase, and UDP-xylose Synthase.
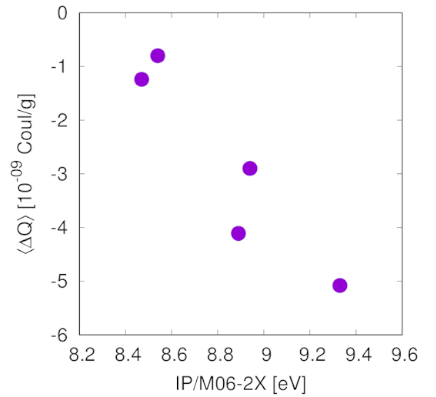
Tribo-charging is a phenomenon that can affect the behaviour of crystalline powders during manufacturing and compaction/tabletting. We measured this effect for various pharmaceutical materials using a GranuchargeTM instrument, and showed that ionization energies, calculated at the DFT level of theory, can efficiently predict tribo-charging trends.